
| TOPICO | MALFORMACIONES DE TEJIDOS BLANDOS BUCALES Y DE LA CARA |
| TIEMPO APROXIMADO | 45 MINUTOS |
| AUDIENCIA |
ALUMNOS CURSO PATOLOGIA ORAL, 3er. año, Fac. Odontología, U Mayor |
| INSTRUCTOR | DR. BENJAMIN MARTINEZ R. |
- I. Racional.
- II. Objetivos Terminales.
- III. Objetivos Específicos.
- IV. Test Inicial.
- Ciclo de Práctica I
- Ciclo de Práctica II
- Ciclo de Práctica III
- Ciclo de Práctica IV
- Ciclo de Práctica V
- V. Test final
I. RACIONAL:
Muchas estructuras orofaciales pueden presentar alteraciones de su desarrollo que se acompañan con otras manifestaciones, a veces muy aparentes,otras no. El dentista en algunos casos puede sospechar una posible asociación, y debe oportunamente derivar el paciente para un óptimo tratamiento.
II. OBJETIVOS TERMINALES:
Podrás describir características clínicas de los síndromes que más frecuentemente afectan la boca.
III. OBJETIVOS ESPECIFICOS:
Cognoscitivos:Estarás capacitado para:a) clasificar síndromes de acuerdo a la herencia mendeliana.
b) enumerar y describir algunas características de síndromes autosómicos dominantes (AD) que afectan los dientes;
c) enumerar y describir algunas características de síndromes que afectan la mucosa oral
d) enumerar y describir algunas características de síndromes que afectan varias estructuras orales.
CICLO DE PRACTICA I:
Generalidades:
Las alteraciones genéticas y a la vez sistémicas, se manifiestan muchas veces en la boca; ya sea porque hay alteraciones en estructuras ectodérmicas o mesenquimáticas. Pueden tener un patrón mendeliano de herencia, aunque eventualmente pueden ser mutaciones y muchas veces tienen un origen multifactorial o poligénico. En la historia clínica es vital para establecer su diagnóstico, construir un árbol genealógico, y con especialistas médicos establecer todas las estructuras, órganos y/o funciones que están alteradas.
Muchas veces una de las principales manifestaciones están en las piezas dentarias, las cuales pueden estar completamente ausentes, tener un desarrollo incompleto, erupcionar en forma tardía, o ser exfoliadas tempranamente. Por esto,en ocasiones, el dentista es llamado a colaborar en el diagnóstico y tratamiento de estos pacientes.
De acuerdo a una clasificación de Gorlin y Sedano (1971), dividiremos estas manifestaciones como:
a) Afectan la mucosa oral:
- Neuromas múltiples de la mucosa, feocromocitoma y carcinoma medular del tiroides (AD).
- Enfermedad de Darier (AD).
- Nevo blanco esponjoso (AD).
- Disqueratosis intraepitelial benigna hereditaria (AD).
- Poliposis gastro intestinal y pigmentación melánica mucocutánea (AD).
- Neurofibromatosis múltiple (AD).
- Fisura labial, palatina (FL(P)), (multifactorial).
b) Afectan los dientes:
c) Afectan varios estructuras orales
- Síndrome nevoide basoceluar AD
- Síndrome oro-facio-digital AR, X-D
- Síndrome Gardner AD
- Síndrome Papillón-Lefévre AR
- Displasia cleidocraneal AD
- Treacher – Collins AD
Observe que al lado derecho se encuentra abreviada la forma de herencia de cada síndrome: autosómico dominante (AD), autosómico recesivo (AR), ligado al sexo dominante (X-D), y ligado al sexo recesivo (X-R).
Referencias Bibliográficas:
- Gorlin, R.J.; Sedano, H.O.: Oral manifestations of systemic disorders. PostGraduate Med. 49:144, 159, 156, 155 (Jan, Feb, Mar, Ap.) 1971.
- Hennekam RCM et al: Gorlin’s syndromes of the Head and Neck. 5th ed. Oxford. 2010.
Retroalimentación
1. ¿Qué es fundamental para hacer el diagnóstico de un síndrome ?
a)
b)
2. Enumere condiciones de origen genético que afectan los dientes
3. ¿Qué alteraciones pueden presentar las piezas dentarias ?
CICLO DE PRACTICA II:
A.- Afectan la mucosa Oral
1.- Neuromas múltiples de la mucosa, feocromocitoma y carcinoma medular de tiroides AD.
Las primeras lesiones en presentarse, en los primeros años de vida son los neuromas que afectan especialmente labios y 1/3 anterior de lengua. El feocromocitoma (tumor de glándula suprarrenal) aparece posteriormente en la pubertad, produce debilidad, palpitaciones, sudoración, náusea, diarrea; probablemente por la producción exagerada de Catecolaminas. En la misma época se presenta el carcinoma de tiroides. Con un diagnóstico precoz del síndrome, pueden evitarse generalmente dichos tumores.
Lesiones hiperqueratinizadas en mucosa oral, vaginal, rectal, laringe, faringe y piel. Inicialmente pápulas rojizas que se fusionan y se tornan grisáceas o parduzcas, de superficie rugosa. Las características histológicas son típicas por formaciones conocidas como cuerpos redondos y granos (que son células epiteliales parcialmente queratinizadas, ubicadas en fisuras del estrato supra basal de la lesión).
3.- Nevo blanco esponjoso. AD.
Lesiones blancas, plegadas, congénitas, de la mucosa oral, generalmente en la mejilla (Fig. 1) y que no afectan el ojo, pero sí a veces con compromiso simultáneo de mucosa vaginal y anal. El frotis citológico es característico por presentar las células epiteliales condensación perinuclear, rojiza, con Papanicolau. Esta no es una lesión blanca cancerizable como otras lesiones de la mucosa bucal que se encuentran en la unidad de lesiones blancas.
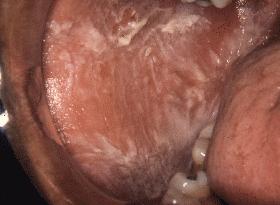
Fig. 1. Cara interna de mejilla derecha donde se aprecian lesiones blancas cubriendo casi la totalidad de ella.
4.- Disqueratosis intraepitelial benigna hereditaria (DIBH)
Lesiones blancas, ásperas de la mucosa oral, asociada a placas gelatinosas de la conjuntiva ocular con base hiperémica, la cual se exacerba en verano y disminuye en otoño, causando ceguera temporal en verano, que se hace definitiva en la 5a.- 6a. década de vida. El frotis es característico, por las células epiteliales célula dentro de otra célula, y células de color anaranjado, alargadas, típicas de observar en frotis teñidos con Papanicolaou. No se ha descrito en Chile.
Gránulos de Fordyce (Condición de Fordyce)
Corresponde a pequeñas pápulas amarillentas que se presentan más frecuentemente en cara interna de mejillas, bilateral, y también a veces en labio superior, en su parte mucosa o borde bermellón. Corresponde a glándulas sebáceas ectópicas, que en piel normalmente se encuentran asociadas a folículo piloso, pero eso no ocurre así en boca. Es muy frecuente en la población, no da sintomatología y no requiere tratamiento, generalmente no presenta ninguna complicación, ni tampoco requiere biopsia.

Fig. 2. Cara interna de ambas mejillas en la misma paciente, con puntos amarillentos.
Referencias Bibliográficas
- Whitten, J.B.; The electron microscopic examination of congenital keratoses of the oral mucous membranes. I. White sponge nevus. Oral Surg. 29:69, 1970.
- Sadeghi, E.M. and Witkop, C.J. : Ultrastructural study of hereditary benign intraepithelial dyskeratoses. Oral Surg. 44:567, 1977.
- Bergsma, D.: Birth defects compendium. 2nd Ed. New York. Alan R Liss. The National Foundation-March of Dimes, 1979, # 352, 339, 681, 538.
- Hennekam RCM et al: Gorlin’s syndromes of the Head and Neck. 5th ed. Oxford. 2010.
Retroalimentación
1. Enumere los signos y síntomas del síndrome neuromas múltiples, feocromocitoma y carcinoma medular del tiroides.
2. En cuanto a los tejidos afectados, ¿qué diferencia hay entre DIBH y la enfermedad de Darier?
3. Una manifestación de la DIBH es:
a) pérdida de la visión en verano
b) pérdida de la visión en invierno
c) pérdida de la visión en otoño
d) pérdida de la visión en primavera
CICLO DE PRACTICA III:
5.- Poliposis gastro intestinal y pigmentación melánica mucocutánea AD.
También conocida como Síndrome Peutz-Jeghers. A veces se presenta poliposis intestinal sin manifestaciones orales con exclusivamente pólipos en colon. En éste caso es mayor la posibilidad de carcinoma el cual puede ser precedido por diarrea con sangre. En el caso que nos preocupa, hay pigmentación color café con leche alrededor de los labios, mucosa de la mejilla y dedos de la mano.
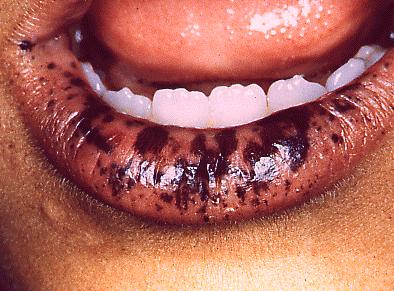
Fig. 3. Niño de 9 años que presentaba múltiples manchas café en labio inferior, borde bermellón y también hacia la mucosa. Posteriormente presentó
6.- Neurofibromatosis múltiple AD.
Síndrome bastante frecuente que se caracteriza principalmente por manchas color café con leche en piel, y tumores cutáneos en relación a troncos nerviosos. Estos tumores (neurofibromas) afectan de preferencia el SNC y tejidos subcutáneo, más graves indudablemente los primeros, que pueden comprometer al nervio óptico, iris; nervios espinales o pueden presentarse otros tumores del SNC (como gliomas o meningiomas).

Fig. 4. Niño con manchas café irregulares en piel del cuello. También presentaba extenso tumor cervical, neurofibroma.

Fig. 5. Neurofibroma mandibular. Note ensanchamiento como trabuco del agujero mentoniano y también deformación en ángulo mandibular, y hacia cóndilo mandibular. La paciente también presentaba múltiples lesiones en piel del tórax y su hija afectada.
7. Fisura labial, palatina (FL(P))
La fisura labial con o sin paladar fisurado, FL(P) es la malformación más frecuente de la cabeza y el cuello y una de las más comunes en el ser humano, además de tener Chile una de las más altas incidencias con un caso cada 650 nacidos vivos. Normalmente se puede encontrar:
- Fisura labial (FL) o también llamado labio leporino.
- Paladar Fisurado (PF)
- Fisura labial unilateral con paladar fisurado
- Fisura labial bilateral con paladar fisurado
De estas la más frecuente es la fisura labial unilateral con paladar fisurado y comprometiendo al lado izquierdo, condición más frecuente en niños hombres. El PF aislado se considera como otra entidad y tiende a ser más frecuente en niñas mujeres. La FL se origina por falta de fusión alrededor de la 6a. o 7a. semana de vida intrauterina de los procesos maxilar y nasal medio. El PF se produciría alrededor de la 8a. semana por falta de fusión de los procesos palatinos, los cuales probablemente en la mayoría de los casos presenten falta de desarrollo.
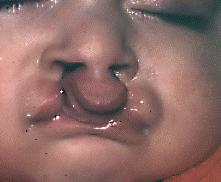
Fig. 6. Fisura labial y palatina bilateral.
La gran mayoría de los pacientes con FL(P) son debidos a herencia multifactorial o sea interacción de factores genéticos y del medio ambiente, que llevan a la fisura. En algunos casos se pueden observar fisuras asociadas a síndromes bien conocidos, con herencia autosómica dominante o de otro tipo, y el riesgo estará de acuerdo a la forma de herencia. En la herencia multifactorial que corresponde a la forma de herencia de la gran mayoría de las fisuras el riesgo en la descendencia es de cerca del 4% y si un padre y un hijo ya han tenido la condición este riesgo aumenta a un 17%. Las complicaciones más frecuentes que se presentan en los niños fisurados se refieren a problemas de deglución, masticación, fonación, estéticos y sicológicos por lo cual el manejo y tratamiento de estos pacientes debe ser realizado por un equipo multidisciplinario, evaluando en el primer examen todas las alteraciones presentes y tomando las medidas preventivas adecuadas para evitar secuelas y complicaciones debido a mal manejo quirúrgico, tratamiento que debe ser controlado y con activa colaboración de los padres del paciente.
 a.
a. 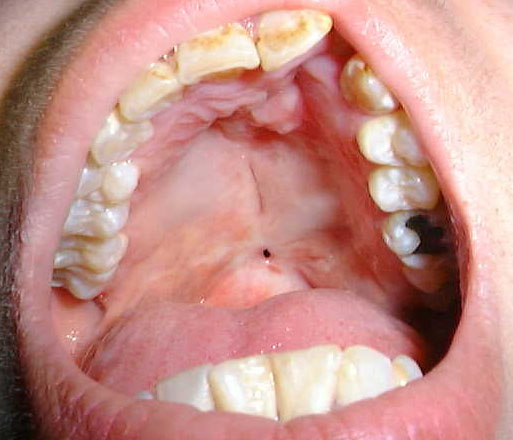 b
bFig. 7. a y b. Paciente operada de fisura labial y palatina. Note secuelas en ala de la naríz del lado izquierdo (a). En vista intraoral (b) se aprecia desviación de central superior, además el lateral no se aprecia, y observe manchas cafesosas en caras vestibulares de dientes anteriores, debidas a hipoplasia del esmalte. También se aprecia pequeña comunicación oro-nasal en paladar blando.
Tabla 1. de Incidencia de Fisuras en Chile y otros países de Latino América (Tasa por 1000 recién nac vivos). Modificada de Nazer et al, Rev Médica de Chile, 129:285-293, 2001.
| País | Fisura labial (palatina) | Fisura palatina |
| Argentina | 1,16 | 0,36 |
| Bolivia | 2,28 | 0,21 |
| Brasil | 1,01 | 0,39 |
| Chile | 1,13 | 0,46 |
| Colombia | 1,03 | 0,30 |
| Ecuador | 1,05 | 0,37 |
| Perú | 0,81 | 0,06 |
| Uruguay | 0,85 | 0,37 |
| Venezuela | 0,77 | 0,33 |
|
1,09 | 0,36 |
Al parecer desde la incorporación de ácido fólico en la harina, desde enero del 2000 en Chile, lo cual también se ha realizado en otros países como USA, Canadá y muchos de
Europa, se ha logrado una disminución de la cantidad de casos de fisurados, o al menos no han seguido aumentado, es probable que la dosis de ácido fólico debiera aumentarse pre concepción de un hijo y durante los primeros meses de embarazo, especialmente en los casos que ya ha habido un niño fisurado.
 Fig. 8. Variación de la tasa de fisurados a partir de 1990 después de iniciada fortificación de la harina hubo un descenso hasta el 2004, posteriormente un incremento debido a que un alto número de molinos NO cumplieron con la norma establecida por el ministerio de Salud.
Fig. 8. Variación de la tasa de fisurados a partir de 1990 después de iniciada fortificación de la harina hubo un descenso hasta el 2004, posteriormente un incremento debido a que un alto número de molinos NO cumplieron con la norma establecida por el ministerio de Salud. Uno de estos síndromes asociado con fisura, colocado en esta página solamente como ejemplo, corresponde al Síndrome de Waardenburg, en el cual además de FL(P) se puede observar hetecromía del iris, sordera sensorineural y alteraciones en pelo y otros tejidos.


Lengua geográfica
También conocida como «glositis migratoria benigna», es una condición que tiene una prevalencia cercan al 10%, y puede tener un origen genético, aunque no se ha demostrado claramente un padrón definido de herencias mendeliana. Puede también ser de origen sicosomático y algunos creen que es una manifestación bucal de psoriasis, o una forma abortiva. En esta condición el dorso de la lengua presenta zonas como si se hubieran perdido papilas, y estas se aprecian enrojecidas, rodeadas por una halo amarillento. En las zonas enrojecidas se están produciendo cambios en papilas filiformes y se aprecian más nítidamente las papilas fungiformes. Estas zonas rojizas con sus halos amarillentos van migrando y de un día a otro se observan cambios en tamaño y aspecto. Histológicamente se encuentra abundantes polimorfonucleares intraepiteliales. No requiere tratamiento pero debiera informarse al paciente e indicarle que es una condición benigna y que no experimenta transformaciones salvo en su aspecto clínico.
Lengua Fisurada
También conocida como «lengua escrotal», es una condición con prevalencia de 7% a 10% y parece ser más frecuente en individuos con retardo mental (Down y otras condiciones). Puede observarse asociada con la enfermedad de Melkersson-Rosenthal. En general se caracteriza por que se observa en el dorso de la lengua, una grieta central de la cual pueden unirse otras grietas que llegan a veces hasta el borde de la lengua y que facilitan el acumulo de restos alimenticios y ocasionar inflamación de ella. Estas grietas pueden presentar gran variación siendo a veces pequeñas y superficiales y otras veces profundas. Cerca del 20% de los pacientes con lengua fisurada también presentan lengua geográfica.
 a.
a. 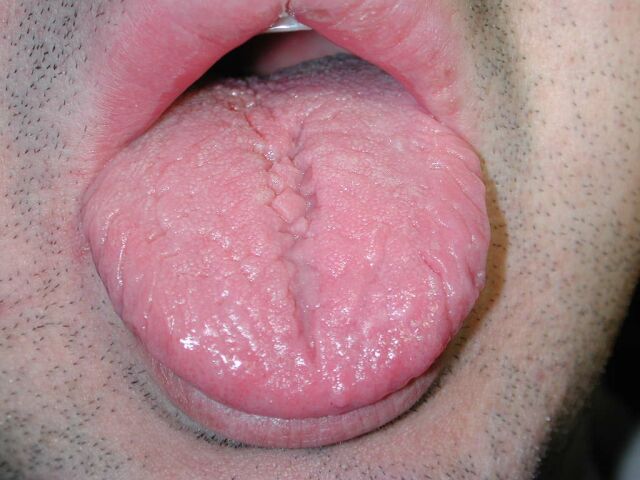 b
b
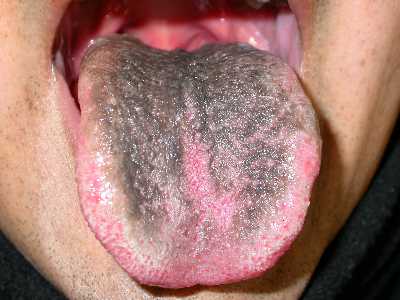
Lengua negra pilosa
Corresponde a hiperplasia de las papilas filiformes debida a sustancias químicas o de causa desconocida. Si está claro que la pigmentación parda o negruzca que se observa en el dorso lingual es debido a pigmentos de origen bacteriano, que posiblemente al quedar numerosas colonias bacterianas entre las papilas descomponen algunas sustancias para ocasionar el color característico que preocupa al paciente y estimula el crecimiento de las papilas filiformes. No requiere tratamiento y solo debiera aconsejarse un enjuagatorio con antiséptico como clorhexidina al 0.1% (aunque nunca debiera usar éste más de 15 días por ser también causante de coloración cafesosa en la lengua por uso prolongado), o cepillado del dorso lingual.
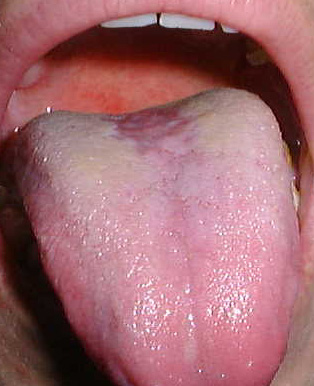
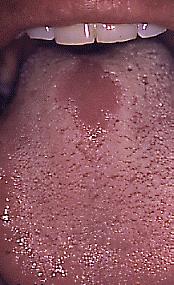
Glositis romboidal media
En realidad no es una malformación, generalmente corresponde a infección con cándida, que ocurre en el dorso de la lengua, por delante de la V lingual, con área depapilada, lisa, generalmente rojiza, pero puede tener zonas blanquecinas (que se desprenden al raspado). En algunos casos se puede observar asociado a VIH, pero también en pacientes portadores de prótesis de acrílico con candidiasis subprótesis. En la biopsia (que debiera postergarse, o no realizarse, ya que debiera tratarse como candidiasis y controlar), se observa epitelio hiperplásico, paraqueratinizado con focos en estrato superficial de neutrófilos (microabscesos), acantosis, papilas elongadas, y corion con infiltrado mononuclear, con degeneración hialina eosinófila en relación a los manojos de fibras musculares. Las hifas de cándida pueden demostrarse en la superficie del epitelio mediante tinción PAS. El tratamiento debiera ser nistatina u otro antimicótico, pero es posible que la mucosa no recupere sus papilas normales, y al parecer esta zona de la lengua, por delante de la V lingual, tiene circulación arterial menor que otras zonas de la lengua, y eso lo puede apreciar en algunos pacientes cuando sacan la lengua, y especialmente al ponerla hacia un costado se aprecia zona isquémica en la misma zona donde se encuentra esta glositis.

Macroglosia
Corresponde a un tamaño grande de lengua que ocasiona que las piezas dentarias impresionen sus caras linguales en el borde de la lengua. Muchas veces la macroglosia es causada por una enfermedad sistémica tal como amiloidosis (no es infrecuente en pacientes con mieloma múltiple); acromegalia; neurofibromatosis; y se observa en el síndrome Beckwith-Wiedemann en la cual también se observa onfalocele (protrusión del intestino por el ombligo), visceromegalia, gigantismo e hipoglicemia neonatal.

Fig. 14. Macroglosia debida a mieloma múltiple. Note cuan marcadas están las caras linguales de dientes en todo el borde lingual, incluso los incisivos.
Anquiloglosia
Es una anomalía del frenillo lingual en que existe una unión fuerte por tejido fibroso y/o muscular entre la lengua y el piso de la boca, a veces llegando a la zona gingival por lingual de los incisivos inferiores, y tiene una prevalencia entre 1 y 7% en la población. La principal complicación es que puede ocasionar alteraciones dentarias en los incisivos inferiores (diastema) y contribuir a la enfermedad periodontal. El tratamiento debe ser la eliminación quirúrgica.

Fig. 15. Frenillo con inserción a nivel de encía lingual de incisivos.
Tiroides lingual
Debido a que la lengua se forma simultáneamente que la glándula tiroides y también a que existe una conexión entre ambas en dicha época, conocido como tracto tirogloso, no es de extrañar que puedan quedar restos de la tiroides en la zona del vértice de la V lingual, que constituyen una especie de nódulo, a veces enrojecido, y que antes de extirpar debiera evaluarse con cintigrafía para no ir a ocasionar un hipotiroidismo, ya que por falla de migración pudiera constituir la parte más importante de la glándula tiroides. Histológicamente este tejido presenta las características normales de la glándula, con sus folículos.
Otros tejidos ectópicos que podemos observar en la lengua son: tejido óseo, formando coristoma óseo, cartílago y en muy raras ocasiones tejido gástrico o intestinal y constituyendo quiste o tumor gastrointestinal heterotópico oral.
B. Afectan a los dientes
Son un grupo de síndromes que afectan el desarrollo de pelos, uñas, glándulas sudoríparas y dientes. En el tipo hipohidrótica, hay incapacidad para controlar la temperatura corporal por la falta de glándulas sudoríparas (hipohidrosis). También presentan estos pacientes escaso pelo en cuero cabelludo, cejas y pestañas (hipotricosis) (Fig. 10); y, en boca hay ausencia parcial de piezas dentarias (generalmente oligodoncia, o sea faltan más de 6 piezas dentarias, en hipodoncias faltan hasta 6), y muchas de las presentes tienen formas anormales, especialmente en incisivos y caninos (formas cónicas) (Fig. 11).
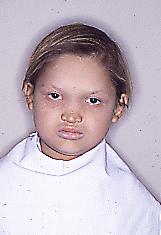
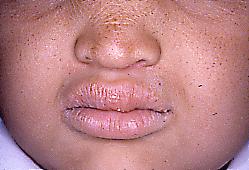
 Fig. 17. Dentición del mismo niño de la foto anterior donde se aprecian los incisivos superiores de formas cónicas, y falta de otros dientes.
Fig. 17. Dentición del mismo niño de la foto anterior donde se aprecian los incisivos superiores de formas cónicas, y falta de otros dientes.
2.- Hipofosfatasia. AR. Falta de la enzima fosfatasa alcalina, que ocasiona una formación ósea anormal, que puede variar desde ocasionar la muerte intrauterina hasta defectos leves en niñez y adolescencia. En los casos de compromiso intermedio, lo más destacado es la pérdida temprana de las piezas dentarias por la falla en el cemento radicular y leves cambios en los huesos. Los hallazgos esqueléticos más frecuentes son cráneosinostosis (cierre temprano de las suturas craneanas), extremidades inferiores arqueadas, tórax pequeño con costillas pequeñas, y deformidad del tipo rosario raquítico. Los enfermos y 60% de portadores del rasgo pueden ser detectados por el aumento de fosfoetanolamina en sangre y orina.
Referencias Bibliográficas
- Witkop, C.J.; Brearley, L.J.; Gentry : Hipoplastic enamel and hypohidrosis inherited as autosomal dominant trait. A review of the ectodermal dysplasia syndromes. Oral Surg. 39(1):71-86, Jan 75.
- Bergsma, D.; Birth Defects Compendium 2nd Ed. New York. Alan R. Liss. The National Foundation March of dimes 1979, Nù 535, 712, 333, 516.
- Holuigue C. Impacto de la fortificación de la harina en Chile en relación a fisuras orofaciales. Trabajo de Investigación, Facultad de Odontología, Universidad Mayor, Santiago, 2005.
- Hennekam RCM et al: Gorlin’s syndromes of the Head and Neck. 5th ed. Oxford. 2010.
- Ver en OMIM. Ectodermal dysplasia hipohidrótica.
Retroalimentación
1. Hipohidrosis se refiere a:
a) falta de uñas
b) falta de pelo
c) falta de dientes
d) falta de glándulas sudoríparas2. Defina anodoncia e hipodoncia
3. La mayoría de los pacientes heterozigotos portadores del rasgo hipofosfatasia pueden ser detectados por la presencia de ……… en……………y………………………
CICLO DE PRACTICA IV:
3.- Raquitismo resistente a Vit. D. X-D.
Síndrome en que hay bajo nivel de fósforo en la sangre (hipofosfatemia), con escasa reabsorción del fósforo inorgánico en túbulos renales. El paciente no responde a las dosis normales de vitamina D usados en el tratamiento del raquitismo. Esta enfermedad puede ser asintomática o acompañarse de osteomalacia o deformaciones raquíticas. Estos últimos casos presentan estatura corta y curvaturas de las piernas, en la boca frecuentemente múltiples abscesos periapicales, debido a que los dientes tienen grandes cámaras pulpares, con finas fisuras que la comunican con el medio bucal. Están afectadas las dos dentaduras (temporal y permanente).
C. Afectan varias estructuras orales
1.- Síndromes nevoide basocelular (SNBC). AD.
Caracterizado por presentar anomalías esqueléticas (costillas bífidas, prominencia del frontal, prognatismo mandibular, puente nasal amplio, Fig. 12); queratoquiste múltiples, los que se estudian en la unidad de Quistes de los maxilares, y carcinomas basocelulares en piel de la cara, cuello y tórax. Además pueden presentar calcificaciones en la hoz del cerebro, y alteraciones oculares.

2.- Síndrome oro-facial-digital.
Existen dos tipos (I y II). El I más frecuente(X-D) con características de: frenillos hiperplásicos, lengua fisurada con dos o más lóbulos, reborde mandibular con fisuras laterales; y, agenesia del incisivo lateral inferior. También presentan fisuras en el paladar; base nasal amplia, hipoplasia del malar y alteraciones en los dedos como clinodactilia (desviación permanente de uno o más dedos); braquidactilia (dedos anormales cortos).
3.- Síndrome Gardner AD. Pacientes que presentan osteomas múltiples en los huesos faciales (frontal, mandíbula); quistes epidermoides y fibromas o desmoides, en el celular subcutáneo. Generalmente se acompaña de pólipos intestinales múltiples, los cuales son de pronóstico reservado ya que experimentan transformación maligna alrededor de los 40 años, si no son extirpados previamente.
Referencias bibliográficas
- Cernéa, P.; Kuffer, R.: Baumant, M.N.; Brocheriou, C. et Guilbert, F.;Naevamatose baso-cellulaire. Association de naevi baso-cellulaire de la Peau deKystes epidermoides des maxillaires, de malformations osseuses et d’aurresanomalies; 7 observations. Rev. de Stomat. 70(3): 181-226, 1969.
- Jones, E.L. and Cornell, W.P. : Gardner’s syndrome; review of the literatureand report on a family. Arch. Surg. 92:287, 1966.
- Hennekam RCM et al: Gorlin’s syndromes of the Head and Neck. 5th ed. Oxford. 2010.
- Síndrome carcinoma nevoide basocelular, OMIM 109400
- Síndrome Gardner, OMIM 175100
Retroalimentación
1. ¿Qué características presenta la dentadura en los niños con raquitismo resistente a vitamina D ?
2. Una anomalía esquelética del síndrome carcinoma névico basocelular es………………; estos pacientes tienen carcinomas basocelulares en la piel de …………………… y…………
3. Hasta este ciclo de práctica hemos nombrado tres poliposis intestinales, ellas son:
a)
b)
c)
CICLO DE PRACTICA V:
4.- Síndrome Papillón-Lefévre, AR.
Este síndrome al igual que la hipofosfatasia y otras condiciones como granuloma eosinófilo, displasia dentinaria radicular, se caracterizan por la pérdida temprana de las piezas dentarias temporales y permanentes en el caso que nos preocupa debido a destrucción del ligamento periodontal, lo cual ocasiona pérdida de la dentadura temporal total a los 3-4 años, y permanente a los 13 – 14 años. Además presentan hiperqueratosis, con enrojecimiento y áreas blancas, en las palmas de la mano y plantas del pie.
5.- Osteogénesis imperfecta, dentino génesis imperfecta AD
Enfermedad compleja que afecta especialmente al esqueleto, también con manifestaciones oculares, cutáneas, otológicas, dentales y vasculares, cuya gravedad es muy variable; en el feto puede ocasionar la muerte por múltiples fracturas y hemorragias intracraneanas. Lo más característico de los pacientes es la gran susceptibilidad a las fracturas, con posteriores deformaciones esqueléticas (extremidades inferiores arqueadas, estatura corta). Cara de forma triangular, con amplia frente. Dientes de color amarillo-cafesosos, opalescente, los cuales pierden fácilmente el esmalte, por alteración en la unión amelo-dentinaria (dentinogénesis imperfecta). Esclerótica azulada y sordera son otras posibles manifestaciones.
6.- Displasia cleidocraneal. AD. Pacientes de corta estatura; se caracterizan por no presentar clavículas. En el cráneo presentan cierre alterado de las suturas, persistiendo abiertas las fontanelas. En la boca tienen el paladar ojival y piezas incluídas, y muchas veces supernumerarios; además falta fusión en la sínfisis mandibular. Presentan otra serie de anomalías esqueléticas en pubis, costillas y vértebras.

 Fig. 20. Facilidad para juntar los hombros, debido a la ausencia o hipoplasia de la clavícula.
Fig. 20. Facilidad para juntar los hombros, debido a la ausencia o hipoplasia de la clavícula.7.- Treacher – Collins (disostosis mandibulofacial) AD.
Pacientes cuyos rasgos más frecuentes son la inclinación hacia abajo de las fisuras palpebrales, coloboma en el párpado inferior (falta de cierre en la fisura ocular embrionaria), arco zigomático poco desarrollado, oídos displásicos; la boca es típica por su micrognacia (mandíbula muy pequeña), que ocasiona alteraciones de la oclusión, y borde basilar cóncavo. Muchas veces puede haber pérdida de la audición, por lo que es imprescindible su diagnóstico oportuno a fin de evitar un retraso mental inducido por la falta de audición.
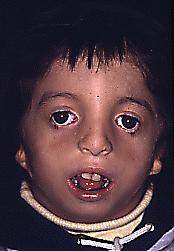 a
a  b
b  c
cFig 21. a, b y c. Niño con Treacher Collins, note inclinación palpebral, falta de desarrollo de maxila y cigoma.
Retroalimentación
1. ¿Qué características presenta el Treacher-Collins?
2. Una anomalía dentaria en la displasia cleidocraneal es ……………….., y a nivel de clavícula puede observarse …………………
Referencias bibliográficas
1.Hennekam RCM et al: Gorlin’s syndromes of the Head and Neck. 5th ed. Oxford. 2010.
2. Síndrome Papillon-Lefevre OMIM 245000
3. Osteogénesis imperfecta, muchos tipos y otros de OI en OMIM.
4. Síndrome Treacher-Collins OMIM 154500
{kind=link}