
| TEMA | PATOLOGIA DEL SISTEMA ENDOCRINO |
| TIEMPO | 45 MINUTOS |
| CURSO | CURSO DE PATOLOGIA DE SISTEMASFacultad de Odontología,U. Mayor |
| INSTRUCTOR | DR. BENJAMIN MARTINEZ R. |
- I. Racional.
- II. Objetivos Terminales.
- III. Objetivos Específicos.
- IV.Test Inicial.
- Ciclo de Práctica I.
- Ciclo de Práctica II
- Ciclo de Práctica III
- Ciclo de Práctica IV
- V. Test Final
I. RACIONAL:
Muchas alteraciones endocrinas puede dar manifestaciones en la cara, y en la boca. El dentista debe reconocer algunas de estas alteraciones y derivar oportunamente un paciente con alteración endocrinológica. .
II. OBJETIVOS TERMINALES:
El alumno podrá reconocer características de algunas alteraciones endocrinas de hipófisis, tiroides, paratiroides y suprarrenal.
III. OBJETIVOS ESPECIFICOS:
El alumno podrá reconocer características de alteraciones:
- Hipofisiarias
- Tiroides
- Paratiroides
- Corteza suprarrenal
- Hiperadrenalismo (Síndrome de Cushing)
- Hiperaldosteronismo
- Hipofunción de la corteza suprarrenal
- Tumores de la cortical suprarrenal.
- Médula suprarrenal
CICLO DE PRACTICA I:
Introducción
La homeostásis es mantenida fundamentalmente por el sistema endocrino y nervioso, los cuales están integrados en el hipotálamo,que regula la función hipofisiaria, y por las células neuroendocrinas ampliamente distribuídas por el organismo. Las hormonas elaboradas por el sistema endocrino interactúan con receptores celulares específicos para cada hormana:
- Hormonas peptídicas y aminas, con receptores sobre la superficie celular, que activan generalmente al AMPc
- Hormonas esteroídeas, atraviesan las membranas celulares, e interactúan con receptores citoplasmáticos e intranucleares. Los complejos hormona-receptor se ligan al ADN y activan genes específicos.
- Hormonas tiroídeas se ligan a la superficie celular y son internalizados para actuar sobre receptores en el interior del citoplasma o, más frecuentemente, al interior del núcleo y también activan genes específicos.
La actividad de algunos órganos endocrinos como la hipófisis es controlada por hormonas estimuladoras o inhibidoras producidas en el hipotálamo. En otras glándulas, como la corteza suprarrenal, las hormonas producidas por la glándula inhiben la síntesis de hormonas tróficas liberadas por el hipotálamo y la hipófisis, proceso conocido como inhibición por retroacción.
Las enfermedades del sistema endocrino se caracterizan por producción excesiva o deficitaria de hormonas con estados clínicos que demuestran hipo ó hiperfunción.
HIPOFISIS
Puede ser por alteración en lóbulo anterior (adenohipófisis) o lóbulo posterior (neurohipófisis), ó en ambos. El aumento anormal de hormonas tróficas (hiperpituitarismo) es causado habitualmente por neoplasia funcional del lóbulo anterior, pero también puede ser por alteración hipotalámica o pérdida de la inhibición por retroacción normal. La disminución de hormonas tróficas (hipopituitarismo) se debe generalmente a destrucción de más del 75% de la adenohipófisis.
Hiperpituitarismo. Tumores del lóbulo anterior.
Es generalmente debido a un adenoma de la hipófisis anterior, y muy raro por alteración del hipotálamo o carcinomas de esta zona. Las hormonas que se producen más frecuentemente son prolactina, hormona del crecimiento y adrenocorticotrofina. Los adenomas de la hipófisis se pueden dividir en microadenomas (<10 mm de diámetro) ó macroadenomas (>10 mm de diámetro). Histológicamente se componen de células monomorfas a diferencia de la adenohipófisis normal. Puede haber:
- Adenomas somatotrópicos. Responsables de la gran mayoría de los casos de exceso de hormona del crecimiento, con acromegalia o gigantismo asociado. La acromegalia se presenta cuando ocurre en los adultos, después del cierre de las epífisis de los huesos, y se presenta aumento de tamaño de la cabeza, manos, pies, mandíbula (la prótesis empieza a quedar chica !). Si el adenoma se presenta antes del cierre de las epífisis se produce el gigantismo, lo cual es bastante raro.
- Prolactinomas. Son los tumores hipofisiarios funcionales más frecuentes y la mayoría son macroadenomas y se caracterizan en el hombre por hipogonadismo y galactorrea (secreción excesiva de la glándula mamaria) en las mujeres.
- Tumores corticotrofos. Son generalmente microadenomas basófilos, y se manifiestan clínicamente con el síndrome de Cushing, por la producción excesiva de ACTH y posterior hipersecreción de cortisol. Los pacientes con este síndrome, generalmente mujeres jóvenes, presentan obesidad, cara de luna, debilidad y fatigabilidad, hirsutismo, hipertensión, intolerancia a la glucosa ó diabetes, osteoporosis y alteraciones neurosiquiátricas. Se tratan con cirugía o dexametasona para disminuir los niveles de ACTH.
Este puede deberse a trastornos de la hipófisis o hipotalámicos, esta últimas siendo muy raras (ej.: craniofaringioma, glioma). Más del 90% de los casos son debidos a trastornos de la hipófisis: adenomas no secretores, el síndrome de Sheehan (infarto de la adenohipófisis) y el síndrome de la silla turca vacía (hernia de la aracnoides y de líquido cefalo raquídeo a través del diafragma hipofisiario con compresión de la hipófisis).. Las manifestaciones clínicas del hipopituitarismo son variables, siendo en el adulto casi indetectable a menos que se evalúe la hormona del crecimiento, en el niño prepúber se ocasiona enanismo hipofisiario.
Referencias Bibliográficas
- Buatti JM, Marcus RB. Pituitary adenomas: current methods of diagnosis and treatment. Oncology (Huntingt) 1997, 11: 791-796.
- Robbins SL, Cotran RS, Kumar V. Manual de Robbins, Patología Estructural y Funcional. McGraw-Hill, Interamericana, Madrid, cap 21, 1997:475.
Retroalimentación
1. ¿Qué tumores de la adenohipófisis conoce ?
2. ¿Cuáles son las alteraciones más frecuentes en el síndrome de Cushing?
CICLO DE PRACTICA II:
TIROIDES
Las alteraciones de la glándula tiroides son relativamente frecuentes en la población. Podemos encontrar: Hipertiroidismo, hipotiroidismo y aumento focal o difuso del tamaño de la glándula.
Es debido a un aumento del nivel de triyodotironina (T3) y de tiroxina (T4) circulantes con un estado hipermetabólico que se manifiesta clínicamente con nerviosismo, pérdida de peso a pesar de aumentar el apetito, piel caliente y húmeda y congestionada por vasodilatación periférica e hipermetabolismo, temblor y aumento de tamaño variable de la tiroides. Las alteraciones oculares son muy llamativas: aumenta la hendidura palpebral, retracción palpebral y en la enfermedad de Graves, proptosis. Las causas más frecuentes, en orden decreciente, del hipertiroidismo son : hiperplasia difusa (Enfermedad de Graves), bocio multinodular tóxico y adenoma tóxico.
Enfermedad de Graves.
Es un hipertiroidismo por bocio difuso hiperfuncionante, con oftalmopatía y dermopatía edematosa (mixedema localizado). Se asocia a HLA-DR3 y otros problemas autoinmunitarios, como tiroiditis de Hashimoto, anemia perniciosa y artritis reumatoídea. Clínicamente existe aumento del tamaño de la tiroides, exoftalmos y dermopatía infiltrativa. A pesar del control de la tirotoxicosis la oftalmopatía puede progresar a una proptosis grave. En el laboratorio se encuentra elevado T3 y T4 y disminuído TSH, y aumento de la captación de yodo radioactivo.
Bocio difuso no tóxico y bocio multinodular.
Generalmente comienza como un aumento difuso del tamaño de la tiroides y posteriormente aparecen los nódulos por lo que se considera la misma enfermedad. En la mayoría de los casos existe una hipertrofia o hiperplasia del epitelio folicular secundaraias a trastorno de la producción de hormonas tiroideas. Existe una forma endémica y esporádica. El bocio endémico tiene mayor prevalencia en zonas con déficit de yodo en la dieta (Alpes, Himalaya, Africa central), pero en muchas partes se ha controlado con el uso de sal yodada (Chile no tiene problema de yodo, algún problema que no tengamos), pero pueden contribuir algunos bociógenos (Calcio, fluoruros, tiocianatos). En el bocio esporádico, menos frecuente, hay una incidencia máxima en la pubertad en adultos jóvenes, pero no se conoce bien como se produce.
Es un estado hipometabólico por déficit de hormonas tiroídeas, que se manifiesta con el cretinismo si comienza en el período perinatal o lactancia, y mixedema en niños mayores y adultos. El cretinismo puede ser endémico por déficit de yodo en la dieta, y se manifiesta con ojos muy separados, edema periorbitario, macroglosia, y dependiendo del inicio grados variables de retraso del desarrollo del esqueleto y cerebral. En el mixedema existe una lentificación insidiosa de la actividad mental, asociada con fatiga, intolerancia al frío y apatía. Pueden haber algunos signos similares a los del cretinismo.
Puede ser de diversas causas, infecciosa por staphylococcus aureus, estreptococos, salmonella, micobacterias u hongos, y de origen hematógeno; autoinmunitario, la tiroiditis de Hashimoto, típico ejemplo de enfermedad autoinmunitaria órgano específico, en la que hay dos tipos clásica y atrófica, y representa la forma más frecuente de hipotiroidismo bociógeno, donde el aporte de yodo es el adecuado. En esta tiroiditis de Hashimoto, existe un hipotiroidismo, muchas veces asociado a aumento de tamaño indoloro de la glándula. Se debe a un problema en la función de los linfocitos T, donde aparecerían células CD4 contra la glándula y producción de autoanticuerpos contra varios componentes: tiroglobulina, receptor de TSH y peroxidasa tiroídea. Parece estar asociada con HLA-DR5, y se presenta asociada con otras enfermedades autoinmunes como Síndrome de Sjögren, artritis reumatoídea, anemia perniciosa.
En USA 2 a 4% de la población presenta nódulos tiroideos solitarios, y la frecuencia es mayor en zonas de bocio endémico. La mayoría de esos nódulos no son neoplásicos, representan quistes, bocio multinodular ó tiroiditis de Hashimoto. Entre las neoplasias más del 90% son adenomas.
Los nódulos presentes en pacientes jóvenes, menores de 40 años, y hombres, tienen mayor probabilidad de ser neoplásicos que los de pacientes mayores.
Los tumores malignos son raros, menos del 1% en USA de las muertes por cáncer. Son más comunes en mujeres y de edad media. Existen distintos tipos:
- Carcinoma papilar (75 a 85%), 3a – 5a. década, generalmente multifocal, invade pronto a los ganglios linfáticos.
- Carcinoma folicular (10 a 20%), 5a – 6a. década, parece que el bocio multinodular predispone a este tipo de neoplasia.
- Carcinoma medular, neoplasia neuroendocrina, secreta calcitonina, estroma de amiloide y puede verse asociado con síndrome NEM (Neoplasia Múltiple Endocrina), este último es más malo el pronóstico que si no está asociado a Síndrome.
Quiste del conducto tirogloso
Anomalía congénita que se presenta como masa en la línea media, entre la tiroides y la base de la lengua.
Referencias Bibliográficas
- Robbins SL, Cotran RS, Kumar V. Manual de Robbins, Patología Estructural y Funcional. McGraw-Hill, Interamericana, Madrid, cap 21, 1997:475.
- Thyroid Disorders. http://www.hsc.missouri.edu/~daveg/thyroid/thy_dis.html
- Thyroid Function tests. http://www.hsc.missouri.edu/~daveg/thyroid/thy_test.html
Retroalimentación
1. Nombre dos tumores malignos de la glándula tiroides y señale una característica de cada uno.
2. ¿Qué es la tiroiditis de Hashimoto?
CICLO DE PRACTICA III:
PARATIROIDES
Hiperparatiroidismo primario (HPTP).
Mas del 90% de los casos de hipercalcemia están relacionados con HPTP o tumores malignos de los huesos. En general el HPTP es debido en la mayoría de los casos a adenoma paratiroídeo (75 a 80%), y el resto a hiperplasia primaria difusa o nodular, o carcinoma de la paratiroides. Puede también observarse en el síndrome NEM. Antiguamente la mayoría de los pacientes se diagnosticaban por la hipercalciuria y los cálculos renales, pero en la actualidad con los exámenes de perfil bioquímico se detectan en general antes y los cálculos renales y las lesiones a los huesos son más infrecuentes. La hipersecreción de paratohormona que existe produce:
- Aumento de la reabsorción ósea y de la movilización de calcio desde los huesos.
- Aumento de la reabsorción tubular renal y de retención de calcio
- Aumento de síntesis renal de 1.25-(OH)2D3 con facilitación de la absorción de calcio por el tubo digestivo
- Hipercalcemia
Hiperparatiroidismo secundario
Es más frecuente en pacientes con insuficiencia renal, pero puede también presentarse en casos de déficit grave de vitamina D u osteomalacia. En los casos de insuficiencia renal se relaciona con la retención de fosfato e hipocalcemia, con hipersecreción compensadora de hormona paratiroídea. En los huesos se presenta la osteitis fibrosa quística y osteomalacia (osteodistrofia renal) con pérdida de las corticales, incluso a nivel de alvéolos dentarios. Si se corrige la insuficiencia reanl peude remitir.
Es una alteración funcional con pocas alteraciones anatómicas. Generalmente existe aumento de la excitabilidad muscular por hipocalcemia y en casos graves puede haber tetania, calambres musculares, estridor laríngeo y convulsiones, alteraciones mentales (irritabilidad, sicosis), calcificación del cristalino y anomalías de la conducción cardíaca. Las causas pueden ser extirpación quirúrgica involuntaria de todas las glándulas paratiroides, ausencia congénita (Síndrome de DiGeorge), destrucción autoinmunitaria.
Referencias Bibliográficas
- Robbins SL, Cotran RS, Kumar V. Manual de Robbins, Patología Estructural y Funcional. McGraw-Hill, Interamericana, Madrid, cap 21, 1997:475.
Retroalimentación
1. ¿Qué es la osteodistrofia renal quística?
2. ¿Porqué un adenoma de paratiroides puede producir hiperparatiroidismo?
CICLO DE PRACTICA IV:
CORTEZA SUPRARRENAL
Hiperfunción (Hiperadrenalismo)
Existen tres síndromes básicos:
- Síndrome de Cushing, exceso de cortisol
- Hiperaldosteronismo
- Síndromes adrenogenitales (exceso de andrógenos)
Como ya habíamos descrito en hipófisis, la hipersecreción hipofisiaria de ACTH puede producir este síndrome, pero también existen otras causas tales como: adiministración de glucocorticoides exógenos (la más frecuente), producción ectópica de ACTH por una neoplasia no endocrina, hipersecreción de cortisol por un adenoma o carcinoma de la suprarrenal o por hiperplasia corticosuprarrenal independiente de ACTH En el 60 a 70% de los casos de síndrome de Cushing existe una hiperplasia corticosuprarrenal difusa, estando afectadas las glándulas a ambos lados y el nivel de ACTH está elevado. Características clínicas del síndrome se describieron en el ciclo de práctica I.

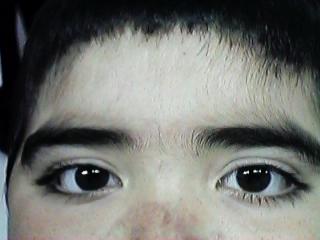
Fig. 1. Niño de 7 años, con facies redondeada, e hirsutismo, debido a la administración de corticoides desde la edad de 2 años. Note la mayor densidad pilosa a nivel de cejas, entre las cejas y en la frente.
Son síndromes raros que se caracterizan por secreción crónica y excesiva de aldosterona independientemente del sistema renina-angiotensina, y que se producen por adenoma solitario secretor de aldosterona (65% de los casos), o hiperplasia suprarrenal.
Síndromes adrenogenitales.
Son manifestaciones raras tales como hermafroditismo, seudohermafroditismo virilización en la mujer y pubertad precoz en el hombre, todos los sindrómes son trastornos autómicos recesivos, con manifestaciones muy variables. El síndrome adrenogenital puede ser causado por neoplasias corticosuprarrenales secretoras de andrógenos ó defectos metabólicos congénitos de la biosíntesis de corticosteroides que determinan una disminución de la producción de cortisol, aumento compensador de la secreción de ACTH, hiperplasia suprarrenal y aumento de otros esteroides de la corteza incluyendo andrógenos.
Insuficiencia corticosuprarrenal crónica primaria (Enfermedad de Addison)
Alteración poco común, de los adultos, que requiere de la destrucción de 90% de la corteza suprarrenal, y que puede ser debida a enfermedad autoinmune de la suprarrenal, infección de la glándula (TBC), o cáncer de ella, pero lo más frecuente es la enfermedad autoinmune.
El paciente con Addison presenta diversas características tales como: debilidad, fatiga, anorexia, hipotensión, náuseas, vómitos e hiperpigmentación, que para el dentista es muy importante, ya que pueden ser iniciales en boca (lengua, mejillas) y que también afectan piel de la cara y en zonas de presión. Los exámenes de laboratorio son necesarios para el diagnóstico y ellos incluyen niveles de ACTH (está elevado), Sodio (bajo), potasio (alto).
Feocromocitoma
La mayoría de las alteraciones de la suprarrenal son neoplasias y la más importante es el feocromocitoma, el neuroblastoma, y ganglioneuroma.
El feocromocitoma es raro y se asocia con la producción catecolaminas e hipertensión, y a veces podría producir síndrome de Cushing u otro trastorno endocrino. Puede ser esporádico o asociado a síndromes especialmente el NEM, aunque también se ha descrito en Sturge-Weber y von Recklinghausen. Este tumor se manifiesta con hipertensión mantenida o persistente y con disfunción de otros órganos como insuficiencia cardíaca congestiva, infarto de miocardio o arritmia cardíaca, y hemorragia cerebral. Las complicaciones cardíacas se atribuyen a la vasoconstricción inducida por las catecolaminas. También puede haber cefalea, sudoración, ansiedad, temblor, trastornos visuales, dolor abdominal y náuseas.
Referencias Bibliográficas
- Robbins SL, Cotran RS, Kumar V. Manual de Robbins, Patología Estructural y Funcional. McGraw-Hill, Interamericana, Madrid, cap 21, 1997:475.
Retroalimentación
1. ¿Qué examen solicitaría a paciente con Addison?
2. ¿Nombre algunas condiciones en las cuales se produce hirsutismo?
Mis opiniones acerca de esta Unidad
¿Algún comentario?, escriba a: