FACULTAD DE ODONTOLOGÍA
UNIDADES DE AUTO –
APRENDIZAJE
|
TEMA:
|
ENFERMEDADES GENETICAS
|
|
CURSO:
|
PATOLOGIA
GENERAL,
2o. año, Odontología, U Mayor.
|
|
TIEMPO
APROXIMADO:
|
45 MINUTOS
|
|
INSTRUCTOR:
|
DR. BENJAMIN
MARTINEZ
R.
|
I. RACIONAL.
II. OBJETIVOS
TERMINALES.
III.
OBJETIVOS ESPECIFICOS.
IV.
TEST INICIAL.
CICLO DE
PRACTICA
I.
- Alteraciones
monogénicas (o enfermedades mendelianas). Como el
nombre
lo indica, solo un gen está alterado, y estas condiciones se
heredan
en forma autosómica o ligada al sexo, dominante o recesiva, de
acuerdo
a las leyes de Mendel. Actualmente existen mas de 3000 enfermedades de este tipo, entre las cuales está la
acondroplasia, algunas anemias hereditarias, anomalías
congénitas del metabolismo, las hemofilias, e incluso algunos
tipos de cáncer. A nivel mundial se cree que entre el1-2% de los
nacidos vivos presentan alguna enfermedad de este grupo. A veces puede
ser el gen alterado una mutación fresca, por lo que no
habrá historia familiar contribuyente, y a veces esto puede ser
debido por la edad avanzada del padre.
- Anomalías
cromosómicas. Se producen ya sea por que existe mucho o poco
material cromosómico, en el primer caso como por ejemplo en el
síndrome
de Down (o mongolismo), y en el segundo caso como en el síndrome
de
Turner. Generalmente se presentan al azar en
hijos
de padres normales. Debe si quedar establecido que existen factores de
riesgo conocidos tales como la edad materna y la exposición
previa a radiaciones. El 50% de los abortos espontáneos presenta
este tipo de alteraciones y se calcula que el 0.6% de los nacidos
vivos, que presentan retardo mental y múltiples malformaciones
congénitas presentan este tipo de alteración.
- Alteraciones
multifactoriales, debidos a la
interacción
de varios genes con el medio ambiente, el cual influye en la
expresión
poligénica. Generalmente se produce una malformación
congénita,
como fisura labial con o sin paladar comprometido, dislocación
congénita
de la cadera y espina bífida o, también una enfermedad
común
del adulto tal como hipertensión, ateroesclerosis y
esquizofrenia.
Las malformaciones congénitas de este grupo afectan entre el
2-3%
de los nacidos vivos y las enfermedades restantes tienen una
prevalencia
general de más del 15% en la población adulta.
Para el reconocimiento de una enfermedad genética, al igual que en cualquier campo de la Medicina, es muy importante la historia familiar Esta historia familiar debe completarse con un árbol genealógico el cual permite determinar algún padrón de herencia mendeliana, si es que hubiera. En segundo lugar es muy importante el examen físico, que debe ser lo mas objetivo posible, buscando cambios estructurales, facies características, alteraciones en las manos, pies, piel, pelo, etc., las cuales deben ser descritas objetivamente. En la actualidad es casi norma en el estudio de una enfermedad, la ayuda de exámenes de laboratorio, que incluye radiografías, estudios bioquímicos, citogenéticos, e inmunológicos, y que en las enfermedades genéticas deben siempre realizarse. Finalmente debe haber un conocimiento previo de los padrones de transmisión genética, la etiología y fisiopatología de la condición y circunstancias que hicieron posible su manifestación para explicárselas al paciente y eventualmente a la familia. También es necesario algo que se ha llamado ojo clínico, o nivel de sospecha mínimo, para estar atento y encontrar formas raras de alteraciones que pueden constituir enfermedad genética y deben ser manejadas correctamente.
Referencias bibliográficas
- OPS.
Prevención
y control de las enfermedades genéticas y los defectos
congénitos.
Informe de un grupo de consulta. Organización Panamericana de la
salud,
Washington, DC, Pub. Científica # 460,1984.
- King
RA. Medical
Genetics. University of Minnesota, Medical School.
Minneapolis,1983.
Retroalimentación
- Qué diferencias hay entre un defecto congénito y uno heredado?
- De un ejemplo de cada uno de los
tipos
de enfermedades genéticas.
CICLO DE
PRACTICA
II
Etiología y prevención de las
Enfermedades
Genéticas
En cuanto al primer aspecto, o sea el grado de
desarrollo
fetal, los agentes que actúan durante la primera semana de vida
intrauterina generalmente se consideran que son
letales, aunque algunos
pudieran sobrevivir y se cree que la trisomía 21 se
originaría
en esta época. En el período de la diferenciación
y
organogénesis (hasta la 9a. semana de vida intrauterina), los
agentes
que actúen provocan severas malformaciones en distintos
órganos,
y entre estas tenemos la fisura labial con o sin
paladar
fisurado (que abreviaremos FLc/s PF). Posteriormente en el
período
fetal (período de la histogénesis y maduración
funcional),
un agente teratógeno (o capaz de producir malformación)
actúa
principalmente en genitales y SNC. En cuanto al estado de la madre,
fisiológico
y patológico, se refiere por ejemplo a la edad de la madre. Es
sabida
la alta frecuencia de anomalías cromosómicas que se
presentan
por la edad avanzada de la madre, así por ejemplo en menores de
35
años la tasa por mil nacidos vivos es de 2, y para mujeres entre
40-44
años se eleva a 25. Por esta razón una de las medidas
fundamentales
en la prevención de estas alteraciones es la de desalentar el
embarazo
en mujeres mayores de 40 años.
El estado patológico de la madre afecta
al
niño en gestación, y también es conocido el
ejemplo
de la rubéola, cuyas lesiones en el niño
dependerán
del tiempo en que se contraiga la enfermedad. Y
para
prevenir esto actualmente se sugiere inmunizar
contra
la rubéola a mujeres en edad reproductiva. Otros agentes
contraídos durante el embarazo se han demostrado que causan
malformaciones como son
la sífilis, diabetes e hipertensión de la madre.
b) La dosis, tiempo y frecuencia de
administración del agente
teratógeno.
Como se señalaba mas arriba, el ejemplo
de
la talidomida ilustra este aspecto. Dicha droga
utilizada
durante la década del 50, como sedante y tranquilizante,
provocó
malformaciones en el 20% de las mujeres que la tomaron, siendo el
período
mas crítico entre la 4-7 semana y siendo suficiente solamente
una
dosis de 50 mg. Las anomalías mas frecuentemente observadas
fueron
amelia (falta de extremidades); focomelia (extremidades poco
desarrolladas);
atresia del esófago, duodeno y ano; aplasia de la
vesícula
y fisura palatina. Otras drogas que se han demostrado como agentes
teratógenos
en humanos son algunos anticonvulsivantes como el fenobarbital,
también
se sospecha en algunos analgésicos que contienen ácido
acetilsalicílico(aspirina),
y finalmente por eso se ha recomendado no ingerir ninguna droga durante
el
embarazo. Hoy en día el agente teratógeno más
importante
en el ser humano es el alcohol, ingerido por la madre por la ingesta de
bebidas
alcohólicas ocasionándose en el feto el síndrome
fetal
alcohólico (en inglés Fetal Alcohol Syndrome), el cual se
caracteriza
por retardo físico y mental y alteraciones faciales (labio
superior
delgado, ausencia de filtrum, pliegues palpebrales cortos).
Indudablemente
que los efectos estarán de acuerdo con la ingesta de bebida
alcohólica,
entre mayor grado alcohólico probablemente más severo el
fenotipo y también en relación a la época del
embarazo en que sea ingerida.
Prevención
- Desalentar
las
gestaciones en mujeres mayores de 40 años.
- Disminuir
los
niveles de exposición a mutágenos y teratógenos
(Agregado
nuestro: toda mujer embarazada debe EVITAR LA INGESTA DE BEBIDAS ALCOHOLICAS y tampoco
debe
FUMAR).
- Detección
prenatal de defectos del tubo neural.
Referencias bibliográficas
- Nanda
R. Teratogenic
effects of envirommental agents on embryonic development. Dent Cl N Am.
19:181-189, 1975.
- OPS.
Prevención
y control de las enfermedades genéticas y los
defectos congénitos. Informe de un grupo de consulta.
Organización
Panamericana de la salud, Washington, DC, pub. científica #
460,1984.
- Malformaciones
Congenitas-
Imágenes, Connecticut University.
Retroalimentación
2. Averiguó para que sirve la
alfa-fetoproteina
en la prevención de algunas malformaciones
congénitas
?
CICLO DE
PRACTICA
III
Alteraciones Monogénicas
Condiciones autosómicas dominantes.
Los
individuos
que presentan una alteración que se hereda en esta forma se
caracterizan
porque todos tienen un padre afectado, elindividuo afectado
generalmente
es heterozigoto, cada hijo de un individuo afectado tiene un 50% de
probabilidad
de heredar el gen, y ambos sexos son afectados
por
igual. Clínicamente, en general, estos desordenes se
caracterizan
porque son rasgos menos severos que los recesivos, tienen
amplia variabilidad de expresión y pueden tener penetrancia
variable. Ejemplosde este grupo son: acondroplasia (enanismo por
extremidades cortas), Síndrome Gardner (pólipos
intestinales con gran tendencia de malignizarse, osteomas y otras
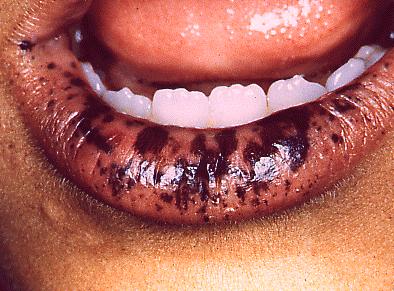
alteraciones), Síndrome Peutz-Jeghers, Fig. 2, (pólipos
intestinales con poca tendencia a malignizarse y manchas
café en boca), disostosis craneofacial o síndrome de
Crouzon,
Fig. 3.

Condiciones ligadas al X recesivas.
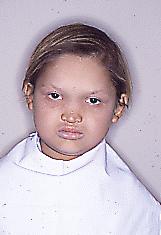
Fig. 4.
Displasia Ectodérmica
hipohidrótica, note ausencia de cejas y
pestañas,
pelo de color claro por falta de pigmento, y surco mentolabial marcado.
Son mas frecuentes estas condiciones en mujeres y las madres heterozigotas transmiten el gen a la mitad de sus hijos varones y la mitad de sus hijas mujeres, pero los hombres afectados lo transmiten solamente a todas sus hijas pero a ningún hijo. Ejemplo de estas condiciones son raros de encontrar en la clínica, uno de ellos es el raquitismo resistente a vitamina D y el síndrome orofaciodigital tipo I.
<>Condiciones ligadas al cromosoma Y.
Durante muchos años se creía que el cromosoma Y solamente transmitía las características sexuales masculinas, hoy se han determinado algunos otros genes en el Y importantes tales como relacionados con la estatura, amelogenina del esmalte dentario, y que tienen en el caso de la mujer un equivalente en el cromosoma X.
Condiciones mitocondriales.
Desde el año 1988 se han descrito algunos trastornos metabólicos y otras alteraciones que se heredan por defectos del ADN mitocondrial, ADN que corresponde al 2% del total de ADN celular, y que se hereda desde el oocito, por lo cual todo ADN mitocondrial es de origen materno. Dentro de estas alteraciones es famoso lo ocurrido con Greg Lemond, notable ciclista, ganador del Tour de Francia, que empezó a desarrollar alteración muscular, miopatía muscular, y tuvo que abandonar el ciclismo. Otras alteraciones mitocondriales son defectos en metabolizar antibióticos (cloranfenicol, estreptomicina), que pueden significar sordera.
Mutación.
La alteración en un sólo gen o
en
un grupo de genes se le llama mutación, y de ella existen varios
tipos: mutación del genoma, o alteración en el
número
de cromosomas; mutación cromosómica o alteración
en
la estructura cromosómicá y, mutación
génica,
o alteración en un solo locus genético. Actualmente se
piensa
que el grado de mutación en células germinativas de
hombres
y mujeres no es igual. Como se recordará la oogénesis
ocurre
durante la vida fetal y la cinética de la
espermatogénesis es
diferente, esto ha conducido a concluir a algunos autores que las
mutaciones del genoma se correlacionan con la edad materna avanzada y
las mutaciones génicas de un solo gen con la edad paterna.
Varios síndromes dominantes como acondroplasia, Síndrome
de Apert y Marfan se han demostrado asociados con edad paterna
avanzada.
Referencias Bibliográficas.
- King
RA. Medical
Genetics. University of Minnesota, Medical School.
Minneapolis,1983.
- Jones
KL, Smith DW, Harvey MA, Hall B & Quan L. Older paternal age and
fresh
gene mutation. J Pediatr 86:84, 1975.
- Salinas C.
(editor).
Genética craneofacial. Organización Panamericana de la
Salud,
Pub. Científica # 378, Washington DC,1979.
- Johns
DR. Mitochondrial DNA and disease. New Engl J Med 333:638, 1995.
- OMIM
Home Page -- Online Mendelian Inheritance in Man.
BASE
DE DATOS DE ENFERMEDADES GENETICAS.
Retroalimentación
2. ¿Qué diferencias existen
entre
expresividad y penetrancia de un síndrome autosómico
dominante.
?
3. ¿Qué condiciones se
transmiten
por las mitocondrias?
CICLO DE
PRACTICA
IV
Anomalías Cromosómicas
Existen cerca de 200 anomalías
citogenéticas
diferentes, numéricas y estructurales y se presentan como hemos
señalado
en el 50% de los abortos espontáneos y el 0.6% de nacidos vivos.
Cuando son muy severas, las que afectan a los
autosomas son letales,
en cambio pequeñas alteraciones de los cromosomas resultan en
severos
cambios fenotípicos.
Anomalías de los cromosomas
autosómicos.
Trisomia 21 o Síndrome Down. Conocido
ya
es el mayor riesgo de presentarse en madres mayores de 35 años.
95%
de los niños con esta condición presentan tres cromosomas
21 (47, XX ó XY, +21). El otro 5%, puede ser
translocación
heredada de un padre portador ó presentarse de novo.
Clínicamente
los niños presentan braquicefalia, pliegues epicantos,
inclinación
mongoloide de los ojos, puente nasal plano, lengua grande y
protruída
con glositis migratoria, defectos cardíacos, corta estatura,
manos
amplias con dedos cortos, hipotonía, retardo mental, y tendencia
a
infecciones pulmonares y leucemia.




Fig. 6. Mujer 35 años con S. Down. Note estrabismo, pliegues epicantos, macroglosia con fisuras (lengua fisurada), dedos cortos, y alteraciones en la palma de la mano.
Anomalías de los cromosoma
sexuales.
Síndrome
Turner. (45,X). Mujeres con cuerpo de Barr negativo. Las
pacientes
con ésta condición presentan bajo peso al nacer, son de
corta
estatura, y tienen baja línea del pelo, pezones muy separados,
falta
de desarrollo sexual, amenorrea primaria, esterilidady
otras alteraciones. Generalmente el X presente es materno.
Síndrome
Klinefelter. (47,XXY). Hombres con
cuerpo
de Barr positivo. Los rasgos clínicos no son aparentes sino
hasta
después de la pubertad, caracterizándose por ser muy
altos
con extremidades inferiores muy largas, testículos
pequeños con hialinización tubular y azospermia,
esterilidad, ginecomastía, y alteraciones pulmonares.
Referencias bibliográficas.
- King
RA. Medical
Genetics. University of Minnesota, Medical School.
Minneapolis,1983.
Retroalimentación
CICLO DE
PRACTICA
V
Alteraciones Multifactoriales
A los 35 días de vida intrauterina el
labio
superior debe haberse fusionado, o sea se han unido los procesos nasal
medio
y nasal lateral de ambos lados, si esto no ocurre habrá
también
una falla, generalmente en el cierre del paladar, el cual no se fusiona
completamente
sino hasta la 8 o 9 semana de vida intrauterina. Otros defectos ocurren
debido
a esta alteración, tales como problemas dentarios, falta de
desarrollo
del ala nasal de ese lado, leve hipertelorismo ocular, alteraciones de
la
masticación, deglución, y fonación, y
también
otitis media por la falta de cierre palatino.
La etiología de esta condición
es
desconocida, puede ser considerada en la mayoría de los casos
poligénica.
La FL c/s PF ocurre más en hombres a diferencia del paladar
fisurado
sólo el que es mas común en mujeres. Entre más
severo
sea el defecto, mayor es el riesgo para la descendencia. La FL
unilateral
tiene un riesgo de 2.7%; pero si es bilateral aumenta a 5.4%.
A veces puede encontrarse fisura, asociada con
síndromes
de herencia mendeliana, donde el riesgo estará de acuerdo al
tipode
herencia que tenga dicha condición; también se ha
observado
en alteraciones cromosómicas.
A veces podemos encontrar tumores
congénitos como Teratoma: malformación tumoral que puede
observarse en boca, ovario u otros tejidos, con formación de
tejidos de distinta hoja embrionaria, o grandes tumores
congénitos, a veces existen antecedentes de marihuana en los
padres (NO fume ni cigarrillos durante el embarazo !).
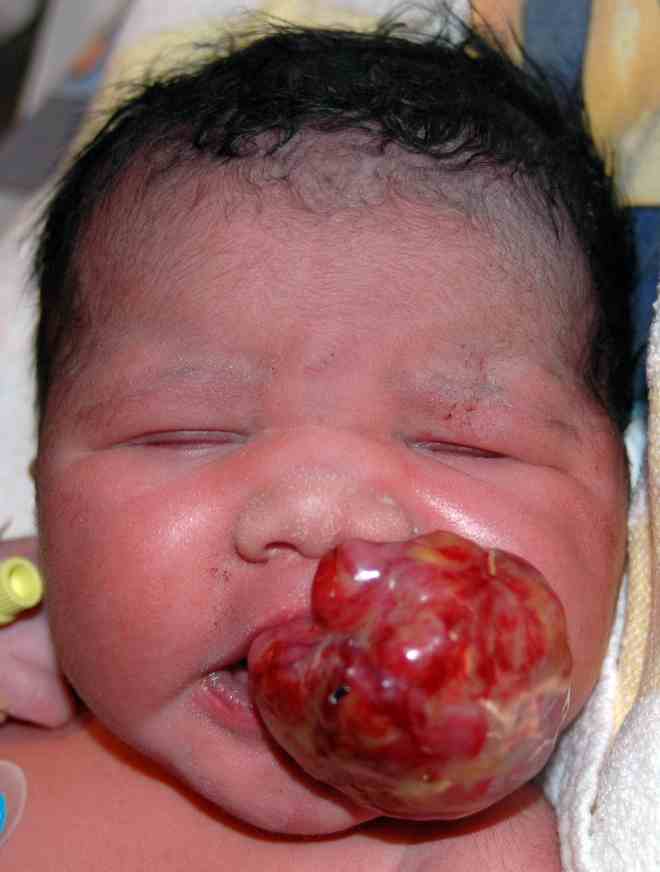
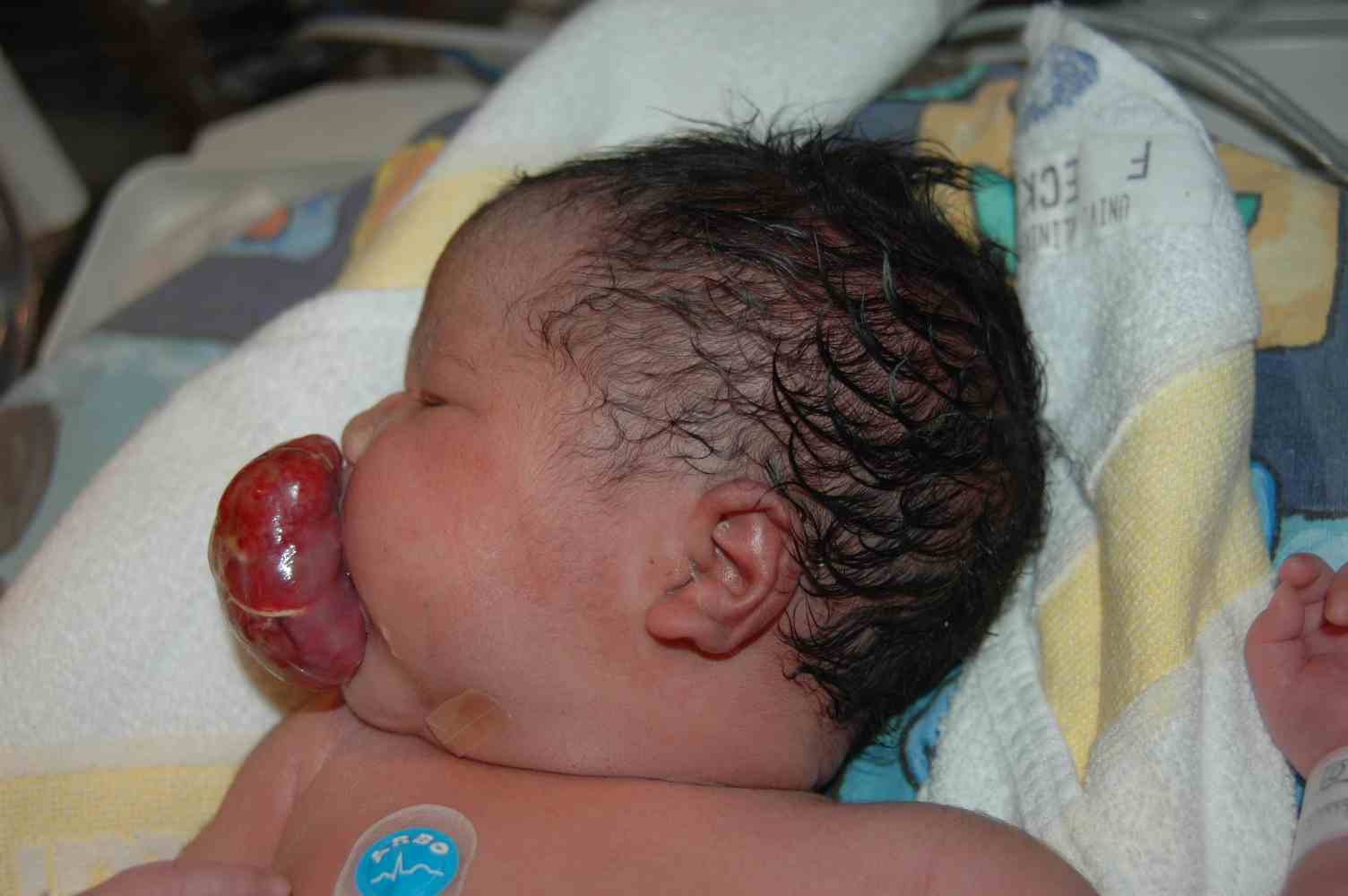
Fig. 6. Epulis congénito del recien nacido, unido a paladar duro. Foto gentileza de Dra. E. Veigel. Caso visto en Alemana y enviado vía correo electrónico, Octubre, 2007.
Referencias bibliográficas.
- Smith
DW. Recognizable
patterns of human malformation, 3a. Ed. Philadelphia,WB Saunders Co.,
1982.
- Gorlin RJ,
Pindborg
JJ y Cohen MM. Síndromes de la cabeza y el cuello.2a. Ed.
Barcelona,
Salvat, 1978.
- Goodman R y
Gorlin
RJ. Atlas of the face in genetic diseases. 2a Ed.
St.Louis,
CV Mosby, 1977.
Retroalimentación.